病例报告表(Case Reprot Form,以下简称CRF)是临床研究中的重要文件,是记录临床资料的方式,被用以记录每一名药物或医疗器械受试者在试验过程中的数据。CRF是临床试验数据采集的必备工具。本文对目前使用最广泛、且被监管部门推荐用于CRF设计的临床数据获取标准协调(Clinical Data Acquisition Standards Harmonization,以下简称CDASH),以及CRF的设计流程进行介绍。
CDASH为CRF设计提供参照
CRF必须包含试验方案所要求的所有字段,这是CRF设计的基本要求。而对于方案要求采集的字段是否合理,以及如何才能在更大程度上保证正确地采集数据,则由CDASH提供参考。
CDASH定义了临床试验数据采集的基本标准,并按数据类型划分数据域,如既往病史(MH)、实验室检查结果(LB)、不良事件(AE)等,并从问题描述、提示、SDTM(Study Data Tabulation Model,试验数据表格样式)或CDASH 变量名称、BRIDG(Biomedical Research Integrated Domain Group)模型、定义、CRF填写指南、申办者补充信息、核心类别8个方面,为数据域中的每个字段提供了统一标准(详见表)。其中,核心类别分为“强烈推荐”“推荐/有条件”“可选”三项。“强烈推荐”字段一般是根据监管部门要求,往往为必选采集字段,对“推荐/有条件”“可选”,可根据情况酌情选择。
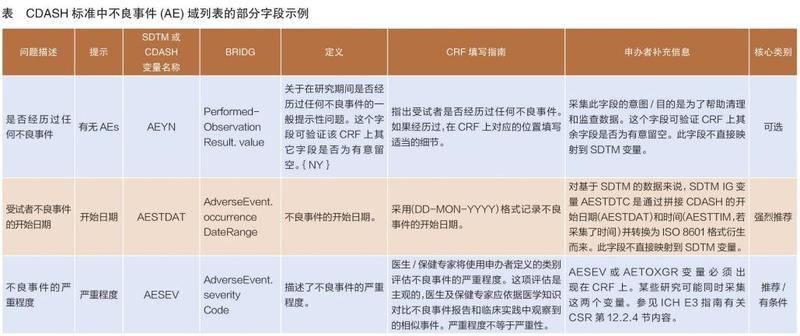
CDASH为数据从采集到递交过程中的转换提供了便利。美国、日本等国家的监管部门均建议在新药注册时按照SDTM递交数据。自我国药品监管部门2017年加入ICH后,我国也逐渐向这一要求靠拢。作为CDISC(临床数据交换标准协会)系列标准中的一环,89%的CDASH变量与SDTM完全相同,为数据的转换提供了极大便利。
由于每个临床试验具有差异性,CDASH只能在最大程度上提供常用数据域及字段标准作为参考,而无法囊括所有的数据列表及试验情况。但由于CDASH对数据域及字段的命名和
定义采用统一规则,所以,当试验中出现新的数据域和字段时,仍可参照其标准进行命名和定义。
CRF设计的一般流程
如果与试验主要评价指标相关的数据点未被采集,试验即可视作失败,因此,CRF设计至关重要。
CRF设计中应有严格的SOP(Standard Operation Procedure,标准作业程序),来对其开发、审核、批准、版本控制及研究中心培训的流程等进行控制。CRF设计员应将包括初稿、审核意见版、定稿及定稿后更新的所有版本进行归档,最终版本由审核各方签字批准。对CRF设计方案的审核应由参与方案设计的人员从各自专业角度(如统计、医学、运营等方向)进行,并将意见统一反馈给CRF设计员。需注意,若定稿后发生更新,应重新进行审核、批准。
CRF的重要性无需再次赘述,但任何完美的方案都不如完美的实施,CRF设计的成功基于对每个数据点的用心把握。
(奥咨达医疗器械服务集团临床研究事业部供稿)



2448d489-197e-40d7-a0c7-bdc2c53eefc0.jpg)
3f585755-a0e7-48d6-b2cd-262e9f1d2ea3.jpg)


